- Disease Overview
- Treatment Overview
Cystinosis is a rare autosomal-recessive lysosomal storage disorder associated with high morbidity, mortality, and a reduced quality of life compared with the general population.1–3
The genetic mutation leads to defective cystinosin function and subsequent intra-lysosomal cystine crystals in almost all tissues and organs in the body, with the kidneys being the most affected organs, resulting in end-stage kidney disease (ESKD) by the end of the first decade of life if left untreated.1,2 Cystinosis is the most common hereditary cause of renal Fanconi syndrome in children.2
Adequate life-long treatment is essential to prevent or to minimise end-organ damage and improve prognosis in patients with cystinosis.1
Incidence
Cystinosis occurs in approximately 1 in 100,000–200,000 live births.1
Higher incidence rate is observed in selected populations with detected founder mutations2 –
The highest birth frequency rate ever reported was in the Pakistani ethnic group living in the West Midlands (UK), with 1 in 3,600 live births affected.2,4
Cause
Cystinosis is caused by bi-allelic mutations in the CTNS gene on chromosome 17 that encodes for cystinosin – a lysosomal cystine-protein co-transporter.1,2
The main impact of this defect is the accumulation of cystine in the lysosomes of affected cells. The low lysosomal pH causes cystine crystal formation which ultimately leads to apoptosis and tissue damage in all organ systems.1,2
Additional roles of cystinosin, beyond cystine transport, that impact cystinosis, include:5
- Proximal tubular cell dedifferentiation
- Impaired vesicular trafficking
- Defective mTOR signalling
- Autophagy
Clinical forms of disease
There are three clinical forms of cystinosis, which can be distinguished depending on the age of presentation and the degree of renal severity, with the below describing how these forms typically manifest without treatment:1,2
Infantile or early-onset nephropathic cystinosis
This is the most frequent and severe form, accounting for 95% of cases. Patients present with renal Fanconi syndrome in the first year of life and in the absence of appropriate treatment, these patients develop ESKD by the end of the first decade of life.1,2 Fanconi syndrome is followed by extra-renal manifestations in untreated disease.2
Juvenile or late-onset nephropathic cystinosis
Patients with this form of cystinosis are diagnosed during late childhood or adolescence, and present with a variable spectrum of features, ranging from isolated asymptomatic proteinuria, a mild renal Fanconi syndrome, to an overt nephrotic syndrome. Progression to ESKD is slower compared with the infantile form of cystinosis, but still occurs between the second and third decade of life.2
These patients develop extra-renal complications at a slower rate compared with those that have the infantile form of cystinosis.2
Adult or ocular cystinosis
The main clinical feature that is observed in patients with this form of cystinosis is photophobia due to corneal cystine accumulation, and this rarely presents before adulthood. The kidneys and other organs are spared from symptoms in most patients with this form of cystinosis.2
Extra-renal complications
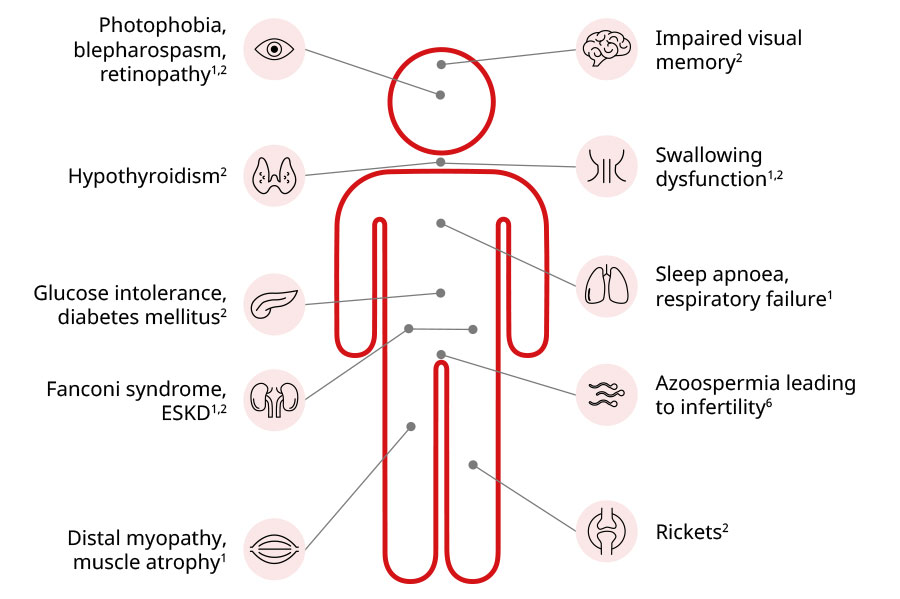
Being a systemic lysosomal storage disorder, cystinosis manifests in almost all tissues and organs. While the build-up of cystine crystals across the body starts early in the course of the condition, the systemic manifestations beyond kidney damage are observed much later.2
Some of the early extra-renal complications seen in patients with cystinosis are rickets and failure to thrive.2,5
Another extra-renal finding is cystine crystal accumulation in the cornea, which usually leads to photophobia and blepharospasm between mid-childhood to early adolescence.2 Deposition of cystine crystals in the cornea may also cause recurrent painful corneal erosions, retinopathy, and lead to blindness.2,5
Systemic complications of cystinosis
Diagnosis2
Blood testing – this is a gold standard diagnostic method which relies on the detection of cystine levels in white blood cells. This method is highly sensitive and precise for detecting the disease.
Genetic testing – can reveal ~95% of disease-causing mutations in the CTNS gene.
Slit lamp examination – used to detect cystine crystals in the cornea.
CTNS, cystinosin; ESKD, end-stage kidney disease; mTOR, mammalian target of rapamycin; UK, United Kingdom.
References:
- Ariceta G et al. Pediatr Nephrol. 2019;34(4):571–578.
- Elmonem MA et al. Orphanet J Rare Dis. 2016;11:47.
- Witt S et al. JIMD Rep. 2023;64(2):199–211.
- Hutchesson AC et al. J Med Genet. 1998;35(5):366–370.
- Cherqui S, Courtoy PJ. Nat Rev Nephrol. 2017;13(2):115–131.
- Reda A et al. Cells. 2021;10(12):3539.
Life-long cystine-depleting therapy with oral cysteamine and cysteamine eye drops are the standard of care for treating patients with cystinosis across the world.1–3
Early initiation of oral cysteamine therapy can improve quality of life and overall disease prognosis. Oral cysteamine therapy delays the onset of end-stage kidney disease (ESKD), and when this complication occurs, kidney transplantation is the treatment of choice.1,2
Cysteamine eye drops are necessary to dissolve cystine crystals in the cornea to ameliorate symptoms such as photophobia, given that oral cysteamine therapy has no effect on corneal cystine crystals.1,2
Cysteamine mechanism of action
Cysteamine enters the lysosome and breaks down cystine into cysteine and cysteine-cysteamine disulfide. These breakdown products are then removed from the cell by an unknown cysteine transporter and the lysine/arginine (PQLC2) transporter.1,4
Oral cysteamine, the only disease-specific therapy for cystinosis, depletes cells and tissues of more than 90% of the cystine content.1–3
Very common and common adverse reactions in patients receiving oral immediate release cysteamine are: Vomiting, nausea, diarrhoea, anorexia, lethargy, pyrexia, Liver function tests abnormal, Headache, encephalopathy, Abdominal pain, breath odour, dyspepsia, gastroenteritis, Skin odour abnormal, rash, Asthenia.
The most common adverse reactions in patients receiving cysteamine eyed-drops are eye pain, ocular hyperaemia, eye pruritus, lacrimation increased, blurred vision or eye irritation. Prescribers should refer to the full list of undesirable effects and special warnings and precautions before prescribing by accessing the prescribing information via the links at the bottom of this page.
Learn more about oral cysteamine and cysteamine eye drops by visiting our resources.
ESKD, end-stage kidney disease; EU, European Union; PQLC2, PQ loop repeat-containing 2; UK, United Kingdom.
References:
- Ariceta G et al. Pediatr Nephrol. 2019;34(4):571–578.
- Elmonem MA et al. Orphanet J Rare Dis. 2016;11:47.
- Nesterova G, Gahl WA. Pediatr Nephrol. 2013;28(1):51–59.
- Cherqui S, Courtoy PJ. Nat Rev Nephrol. 2017;13(2):115–131.
IE-NPS-0112 | June 2026