- Disease Overview
- Treatment Overview
Acromegaly and Cushing’s Syndrome (CS) are endocrine disorders associated with a broad range of physical and clinical manifestations, as well as with high morbidity and mortality if left untreated.1–4
Acromegaly results from excessive production of growth hormone (GH) from a pituitary adenoma, which increases secretion of insulin-like growth factor 1 (IGF-1) and leads to overgrowth of several tissues in the body.1,2
CS (also known as hypercortisolism) results from prolonged exposure to excessive levels of cortisol. Exogenous hypercortisolism is the most common form of CS and is caused by prolonged use of corticosteroids.3,4
Endogenous CS results from excessive production of cortisol by the adrenal glands and can be ACTH-dependent or ACTH-independent.3
Cushing’s disease (CD) is caused by an ACTH-secreting pituitary adenoma and is the most common form of endogenous CS.3,4
Incidence
The incidence of acromegaly is 2–11 cases per million annually5
The incidence of CS is 1.8–4.5 cases per million annually6
Acromegaly cause
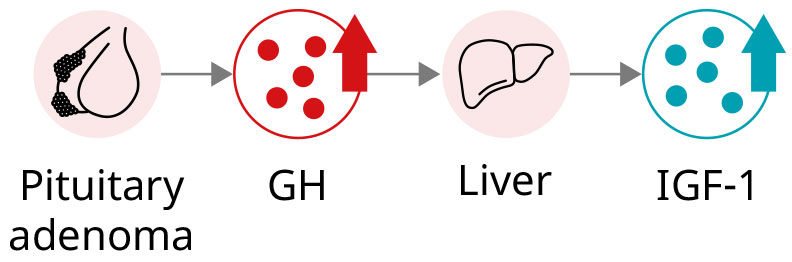
Most cases (>95%) of acromegaly are caused by chronic hypersecretion of GH from a pituitary adenoma, which consequently leads to hypersecretion of IGF-1.1,7
CS cause
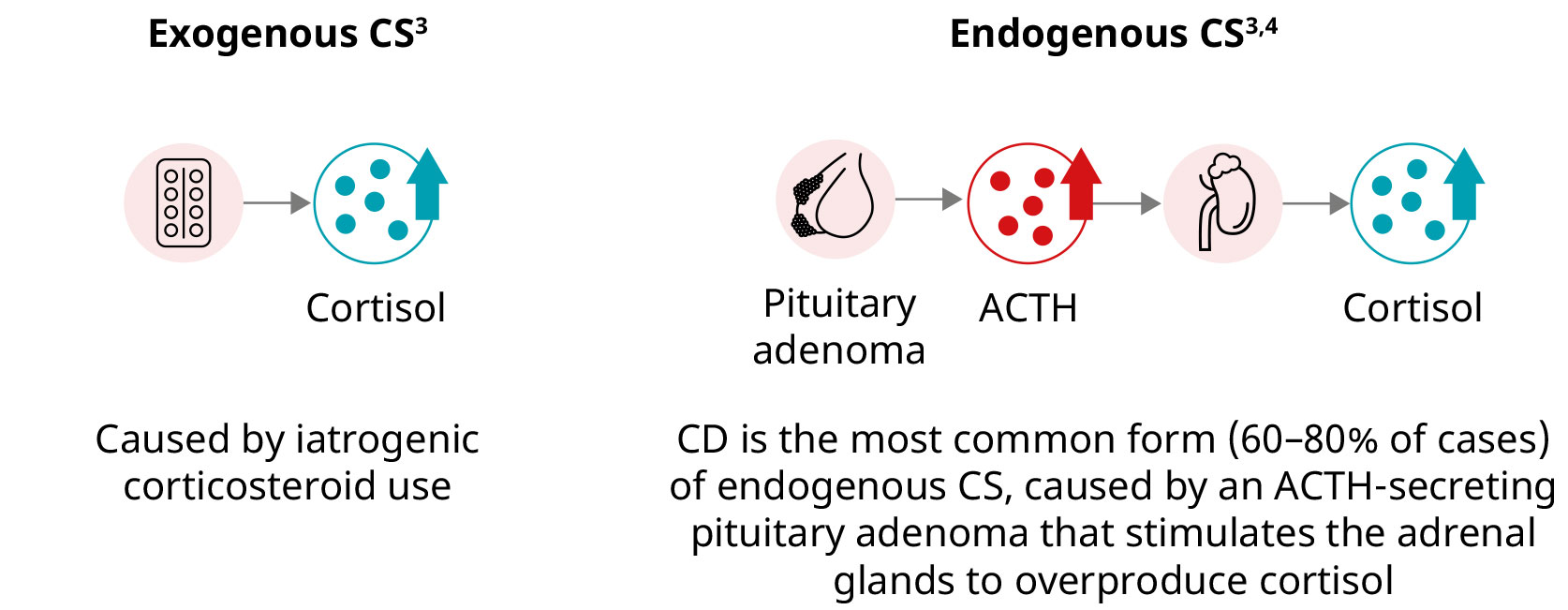
CS is caused by long-term exposure to high circulating levels of the hormone cortisol. CS is divided into two main types based on whether the source of excessive cortisol is external (exogenous CS) or within the body (endogenous CS).3,4
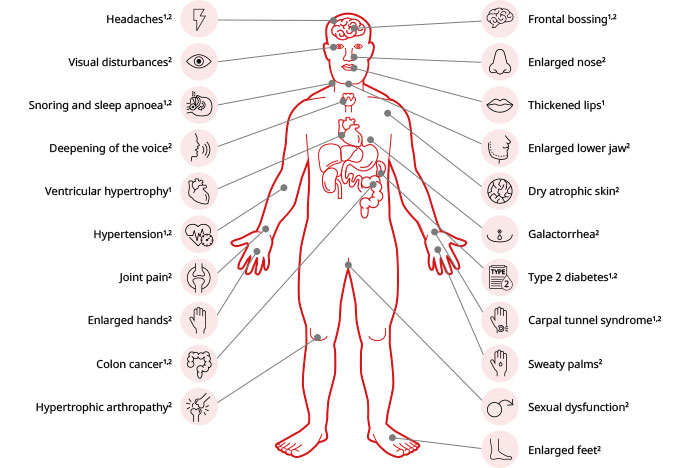
Signs, symptoms, and potential complications of acromegaly
Signs, symptoms, and potential complications of CS
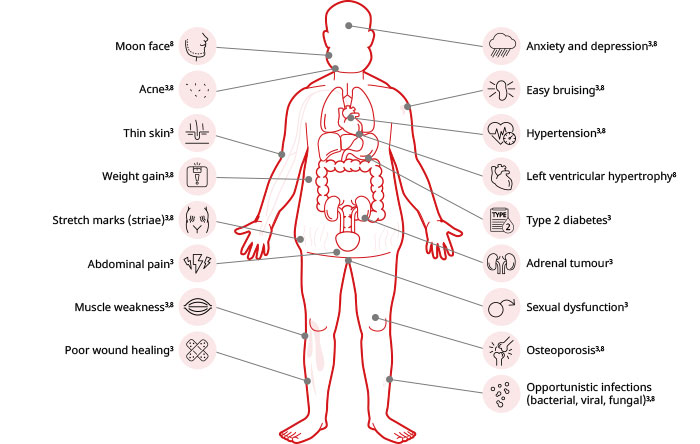
Acromegaly diagnosis
The broad range of manifestations and similarity to other more common conditions makes diagnosis of acromegaly challenging and can lead to a mean delay of ~10–11 years in diagnosing this condition.9,10
Measurement of serum IGF-1 is the diagnostic test of choice, being recommended in patients showing typical clinical manifestations of acromegaly, as well as in those without typical manifestations who show several associated complications1,7
A GH suppression test (before and after glucose administration) is recommended to confirm diagnosis in symptomatic patients with equivocal serum IGF-1 levels2,7
Imaging studies of the brain (using MRI or CT scans) are recommended to locate and visualise the tumour1,2,7
CS diagnosis
The broad range of manifestations and variable symptoms among patients with CS makes diagnosing this condition challenging, with patients experiencing an average delay to diagnosis of ~4 years.8,11
The first step in the diagnosis of CS is to exclude corticosteroid use. Subsequently, it is recommended to conduct one of the following first-line screening tests:8
Overnight 1-mg dexamethasone suppression test (DST)
24-hour urinary free cortisol*
Late-night salivary cortisol test*
Abnormal test results should prompt further evaluation by an endocrinologist, and patients should be tested again with one or two first-line screening tests or a second-line screening test (e.g. 48-hour 2-mg DST) to establish a diagnosis of CS.8
*This test should be performed at least twice due to significant day-to-day variations in cortisol production.8
ACTH, adrenocorticotropic hormone; CD, Cushing’s disease; CS, Cushing’s syndrome; CT, computed tomography; DST, dexamethasone suppression test; GH, growth hormone; IGF-1, insulin-like growth factor 1; MRI, magnetic resonance imaging.
References:
- Ershadinia N, Tritos NA. Mayo Clin Proc. 2022;97(2):333–346.
- Adigun OO et al. Treasure Island (FL): StatPearls Publishing. 2024.
- Chaudhry HS, Singh G. Treasure Island (FL): StatPearls Publishing. 2024.
- Reincke M, Fleseriu M. JAMA. 2023;330(2):170–181.
- Lavrentaki A et al. Pituitary. 2017;20(1):4–9.
- Gadelha M et al. Lancet. 2023;402(10418):2237–2252.
- Katznelson L et al. J Clin Endocrinol Metab 2014;99(11):3933–3951.
- Savas M et al. J Clin Endocrinol Metab. 2022;107(11):3162–3174.
- Caron P et al. Endocrine. 2019;63(1):120–129.
- Zahr R, Fleseriu M. Eur Endocrinol. 2018;14(2):57–61.
- Kreitschmann-Andermahr I et al. Eur J Endocrinol. 2015;172(3):285–289.
The treatment goals of acromegaly and Cushing’s Syndrome (CS) include normalisation of biochemical parameters, associated symptoms, managing comorbidities, and improving survival.1–4
The treatment of choice for both acromegaly and endogenous CS is surgical removal of the tumour. 1–6 For patients with exogenous CS, the best therapy involves tapering corticosteroids gradually.5
Medical therapy is recommended as a second-line option for the treatment of acromegaly, with radiotherapy being a third-line intervention.1–3 For the treatment of endogenous CS, several second-line therapeutic options exist, including repetition of surgery, radiotherapy, and medical therapy.4–6
Treatment for acromegaly
Treatment goals include:
- Normalisation of growth hormone (GH) secretion1–3
- Normalisation of serum insulin-like growth factor 1 (IGF-1) levels1–3
- Symptom improvement/resolution1,2
- Managing associated comorbidities1,2
- Improving survival1,2
Patients with acromegaly whose disease is controlled (i.e. normalisation of GH/IGF-1 levels) have a similar life expectancy to the general population1,2
Surgery
Transsphenoidal surgery (TSS) to remove the pituitary tumour represents the standard of care for most patients.1,3
When conducted by experienced surgeons, TSS results in an initial remission rate up to 90% for microadenomas and 40–60% for macroadenomas.1,3
Medical therapy
Medical therapy is recommended as a second-line option for patients who do not achieve surgical remission, those who are not candidates for surgery, or patients who want to avoid surgery.1–3
Treatment with a somatostatin receptor ligand (SRL) or the GH-receptor antagonist pegvisomant are recommended for patients with significant disease activity (i.e. moderate-to-severe signs and symptoms of GH excess and without local mass effects).3
Medical therapies for acromegaly1–3
Radiotherapy
Radiotherapy is recommended as a third-line treatment option in patients who fail to achieve remission after surgery, and when medical therapy is ineffective, not tolerated, or unavailable.1–3
Patients treated with radiotherapy need to be closely monitored for hypopituitarism and delayed radiation effects.1–3
Treatment for CS
Treatment goals include:
- Normalisation of cortisol levels4
- Symptom improvement/resolution4
- Managing associated comorbidities4,5
- Improving survival5,6
Exogenous CS
The treatment of choice for exogenous CS is to taper exogenous corticosteroids gradually over time to allow adrenal function to recover, which can take several months5
Surgery (endogenous CS)
Surgery to remove the adrenocorticotropic (ACTH) secreting tumour, or adrenalectomy, represents the first-line treatment intervention for patients with endogenous CS.4–6
In Cushing’s disease, TSS results in remission rates up to 90% for microadenomas and <65% for macroadenomas.6
Unilateral adrenalectomy is curative in nearly 100% of adults and children with cortisol-producing adrenal adenomas when conducted by experienced surgeons.4
Second-line treatment options (endogenous CS)
When surgery is unsuccessful or not possible, the choice of second-line therapy should be based on several factors, including treatment goals, urgency to treat, size and location of residual tumours, patient preferences, side effects, and availability of medical interventions4
Bilateral adrenalectomy is recommended in patients with occult or metastatic ectopic ACTH syndrome (EAS), or as a life-preserving emergency treatment in patients with severe ACTH-dependent disease that cannot be controlled with medical therapy4
TSS may be repeated in patients with evidence of incomplete resection or a pituitary lesion showing on imaging4,6
Radiotherapy/radiosurgery is a treatment option in patients who have failed surgery and in those with recurrent Cushing’s disease (CD)4–6
Medical therapy may be used as an initial approach in patients with: ACTH-dependent CS who have failed surgery, those with persistent metastatic disease, or with an occult tumour4
Medical therapy (endogenous CS)
Medical therapies either target the central inhibition of ACTH-secreting adenomas, the adrenal inhibition of steroidogenesis, or the glucocorticoid-receptor blockade6
Treatment with steroidogenesis inhibitors is recommended following TSS in patients with CD; as primary treatment of EAS in patients with occult or metastatic disease; and as adjunctive treatment to reduce cortisol levels in adrenocortical carcinoma4
Pituitary-directed treatments are recommended in patients with CD who are not candidates for surgery or have persistent disease after TSS4
Medical therapies for endogenous CS4,6
About Signifor® (pasireotide)
Signifor (pasireotide) is indicated in the UK and the EU, for the treatment of adult patients with acromegaly for whom surgery is not an option or has not been curative and who are inadequately controlled on treatment with another somatostatin analogue.7
Signifor is also indicated in the UK and the EU, for the treatment of adult patients with Cushing’s disease for whom surgery is not an option or for whom surgery has failed. The 60 mg strength is only to be used in the treatment of acromegaly.7
Isturisa (osilodrostat)†
Isturisa (osilodrostat) is indicated in the UK & EU for the treatment of endogenous Cushing’s syndrome in adults.8
Learn more about Signifor by visiting our Resources
†Not subject to additional monitoring in Ireland.
ACTH, adrenocorticotropic hormone; CD, Cushing’s disease; CS, Cushing’s syndrome; EAS, ectopic ACTH syndrome; EU, European Union; GH, growth hormone; IGF-1, insulin-like growth factor 1; SRL, somatostatin receptor ligand; TSS, transsphenoidal surgery; UK, United Kingdom.
References:
- Ershadinia N, Tritos NA. Mayo Clin Proc. 2022;97(2):333–346
- Adigun OO et al. Treasure Island (FL): StatPearls Publishing. 2024.
- Katznelson L et al. J Clin Endocrinol Metab. 2014;99(11):3933–3951.
- Nieman LK et al. J Clin Endocrinol Metab. 2015;100(8):2807–2831.
- Chaudhry HS, Singh G. Treasure Island (FL): StatPearls Publishing. 2024.
- Kairys N et al. Treasure Island (FL): StatPearls Publishing. 2024.
- Signifor (pasireotide pamoate) Summary of Product Characteristics.
- Isturisa (osilodrostat phosphate) Summary of Product Characteristics.
IE-NPS-0115 | June 2026