- Disease Overview
- Treatment Overview
Neuroblastoma is the most frequently occurring extracranial solid tumour of childhood, accounting for 8–10% of all paediatric cancers and up to 15% of cancer deaths in children.1–3
Neuroblastoma is a tumour originating from neural crest progenitor cells, that can occur anywhere within the sympathetic nervous system, including the superior cervical, paraspinal, and celiac ganglia; with most tumours arising in the adrenal medulla.2,3
Neuroblastoma is a highly heterogenous disorder, demonstrating high variability in its clinical presentation and disease progression, with some children having tumours that regress completely without treatment, while others present with major illness and widespread metastatic tumours with poor survival outcomes.1,2
Incidence
The estimated incidence of neuroblastoma is ~10.2–10.5 cases per million children under the age of 15 in Europe and North America.1
Cause
Neuroblastoma can develop sporadically or be transmitted in the germline, but the exact cause of this condition remains unknown.1,2
~1–2% of all neuroblastoma cases are associated with a positive family history, displaying autosomal dominant inheritance with incomplete penetrance.1
Amplification of the MYCN oncogene is seen in ~25% of patients and is associated with poor prognosis.2
Several maternal risk factors have been associated with the development of neuroblastoma:
Older maternal age5
Maternal hypertension4
Maternal gestational anaemia5
Maternal gestational alcohol consumption6
Risk stratification
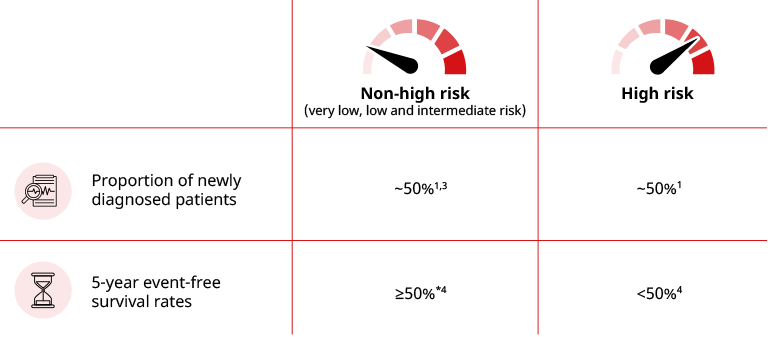
The large biological and clinical heterogeneity in neuroblastoma has been linked to multiple factors associated with patient prognosis. These prognostic factors include:1,2
Clinical characteristics – tumour stage and patient’s age at diagnosis1,2
Biological features – tumour histology and DNA ploidy1,2
Cytogenetic factors – amplification of the MYCN oncogene and key chromosomal gains or deletions1,2
Serum tumour markers – such as ferritin and lactate dehydrogenase (LDH)1,2
Using subsets of known prognostic factors, patients with neuroblastoma can be categorised into two main risk groups:1,3

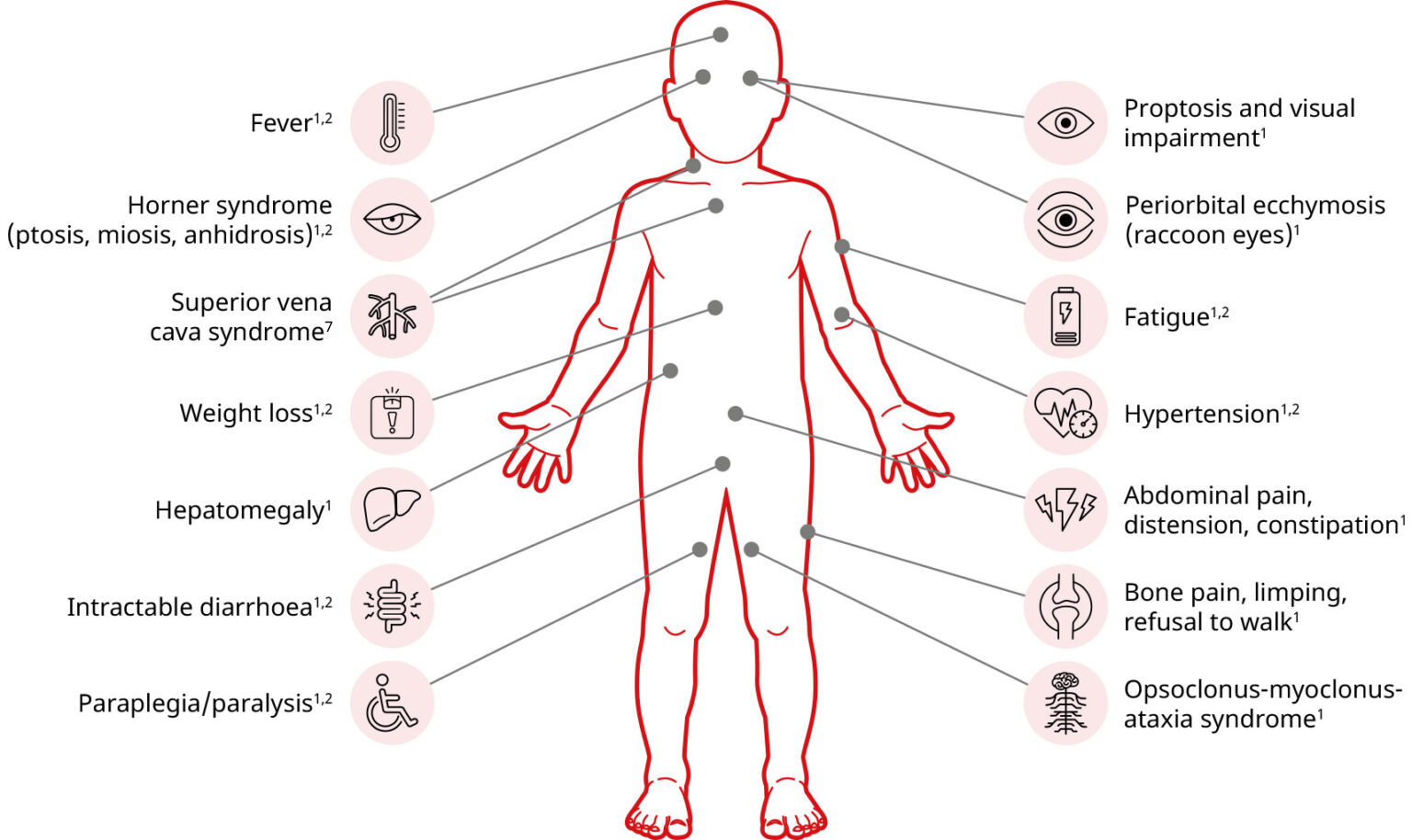
Potential signs, symptoms, and complications of neuroblastoma
References:
- Whittle SB. Expert Rev Anticancer Ther. 2017;17(4):369–386.
- Mahapatra S, Challagundla KB. Treasure Island (FL): StatPearls Publishing. 2024.
- Meany HJ. Children (Basel). 2019;6(1):5.
- McLaughlin CC et al. Cancer Causes Control. 2009;20(3):289–301.
- Bluhm E et al. Int J Cancer. 2008;123(12):2885–2890.
- Heck JE et al. Paediatr Perinat Epidemiol. 2009;23(2):125–143.
- Ozcan A et al. North Clin Istanb. 2020; 7(3):255–259.
- American Cancer Society. Available at: https://www.cancer.org/cancer/types/neuroblastoma/detection-diagnosis-staging/how-diagnosed.html. Date accessed: January 2026.
Neuroblastoma treatment and intensity varies substantially depending on patient risk group (low, intermediate, or high), ranging from observation alone for some low-risk patients, to multi-modal aggressive therapy involving surgery, chemotherapy, radiotherapy, and immunotherapy in high-risk patients.1–4
Children with high-risk neuroblastoma represent a highly challenging group, with the treatment approach being complex and divided into three main phases: commencing with induction chemotherapy and tumour resection; followed by consolidation with high-dose chemotherapy with autologous stem cell transplantation (ASCT) and radiotherapy; and post-consolidation with anti-disialoganglioside 2 (GD2)-based immunotherapy and isotretinoin.4
Treatment outcomes for children with non-high-risk neuroblastoma (low- and intermediate-risk categories) are excellent, with long-term event-free survival (EFS) and overall survival (OS) rates being >90%, whereas children with high-risk neuroblastoma experience EFS and OS rates of <50% despite the use of aggressive multimodal therapy.1–4
High-risk neuroblastoma treatment
The treatment of high-risk neuroblastoma is divided into three main phases (induction, consolidation, and maintenance), lasting for approximately 18 months.4,5
Treatment phases of high-risk neuroblastoma
| Induction |
| Aim: To reduce the size of the primary and/or metastatic tumour and collect stem cells in preparation for ASCT4,5 |
| Interventions: Chemotherapy, surgical resection, stem cell collection4,5 |
| Consolidation |
| Aim: To eliminate any remaining minimal residual disease or gross disease4,5 |
| Interventions: High-dose chemotherapy with ASCT, radiotherapy4,5 |
| Maintenance |
| Aim: To treat residual disease that remains despite induction and consolidation treatment1,5 |
| Interventions: Anti-GD2 immunotherapy, isotretinoin4 |
ASCT, autologous stem cell transplantation; EFS, event-free survival; GD2, disialoganglioside 2; IgG, immunoglobulin G; OS, overall survival.
References:
- Whittle SB. Expert Rev Anticancer Ther. 2017;17(4):369–386.
- Mahapatra S, Challagundla KB. Treasure Island (FL): StatPearls Publishing. 2024.
- Meany HJ. Children (Basel). 2019;6(1):5.
- Krystal J, Foster JH. Children (Basel). 2023;10(8):1302.
- Smith V, Foster J. Children (Basel). 2018;5(9):114.
- Zage PE. Children (Basel). 2018;5(11):148.
- Qarziba (dinutuximab beta) Summary of Product Characteristics.
IE-NPS-0117 | June 2026